Original article from JAMA Network
The search for biomarkers in Alzheimer disease (AD) has been driven by the expectation that such markers will facilitate diagnosis in advance of significant clinical impairment, as well as serve as surrogate markers for clinical trials. The leading biomarkers now in practice are directed at early identification of the 2 neuropathologic hallmarks of the disorder, namely, amyloid plaques and neurofibrillary tangles. The widely defended view is that these pathologic changes silently accumulate in the brain over years if not decades before symptoms occur. Therefore, having meaningful biomarkers would have the potential to shape diagnosis, disease-modifying therapies, and preventive measures in the future. However, the definition of disease through biomarkers rather than symptoms can lead to confusion.
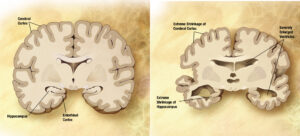
Image courtesy of Wikipedia
For example, consider a 50-year-old man with a sedentary lifestyle, documented hyperlipidemia that is only moderately controlled by diet, and moderate hypertension for which he is poorly adherent to prescribed medications. Because he does not participate in any strenuous activities, always opts to drive rather than walk, and takes the elevator rather than climb stairs, he has not experienced symptomatic angina. If he underwent coronary angiography, it would be expected that atherosclerosis involving multiple coronary artery territories would be documented. At autopsy for an unrelated cause, coronary artery atherosclerosis would not be a surprise to the pathologist. From either diagnostic approach, it would be routine to establish a diagnosis of atherosclerotic coronary artery disease, without concern as to whether he had been symptomatic with the typical manifestations, such as angina. This scenario highlights the need to use distinct language to describe disease processes as reflected in pathologic and biomarker findings vs clinical symptom complexes (coronary artery atherosclerosis vs angina).
Unfortunately, the historical intermingling of clinical neurology and neuropathology has commonly masked such distinctions for neurologic diseases, as is well exemplified by the description more than 100 years ago by Alois Alzheimer of both the clinical syndrome and the histologic hallmarks of the disorder that bears his name. A National Institute on Aging–Alzheimer Association (NIA-AA) workgroup has recently proposed a research framework in which AD is defined by neuropathologic or biomarker evidence of amyloid and tau lesions regardless of the presence or absence of clinical symptoms.1 The hypothetical benefit is to assist in identifying asymptomatic individuals who are at the greatest risk of developing clinically manifest AD to intervene before irreversible damage has occurred in their brains. The challenge is to accurately predict who will develop symptoms and when, if ever, they will appear. For diseases that most commonly affect older adults, there is the complicating association of other processes that determine life span. However, when faced with a similar conundrum of relating neuropathologic findings to symptomatic disease, the neuropathologic community opted for the terminology of Alzheimer disease neuropathologic changes because of the expected mismatch between lesions and symptoms.2,3
In this issue of JAMA Neurology, Jack and colleagues4 report their evaluation in 3 nested cohorts of the Mayo Clinic Study of Aging (MCSA) of sex- and age-specific prevalence of the following 3 imaging biomarker–based definitions of the AD spectrum from the NIA-AA research framework: Alzheimer continuum (abnormal amyloid regardless of tau status), Alzheimer pathologic change (abnormal amyloid but normal tau), and AD (abnormal amyloid and tau). They compared them with the prevalence of 3 clinically defined diagnostic entities commonly linked to AD, including mild cognitive impairment or dementia, dementia, and clinically defined probable AD. The authors found that biologically defined AD is more prevalent than clinically defined probable AD at any age and is 3 times more prevalent at age 85 years in men and women. Because this difference is mostly driven by asymptomatic individuals with imaging biomarker–defined AD, they suggest that these findings illustrate the magnitude of the potential consequences on public health by intervening on asymptomatic individuals with positive AD imaging biomarkers.
Jack and colleagues4 have carefully studied participants in the MCSA cohort who underwent amyloid positron emission tomography (PET) (n = 1524) or both amyloid and tau PET (n = 576) along with longitudinally assessed cognition. This large sample is particularly informative because, in addition to its size, it is a population-based and not a clinic-based cohort. However, their study also represents key challenges that need to be addressed before imaging biomarker–based definitions of AD can be implemented in clinical settings, although such definitions are already being used in clinical trials conducted in asymptomatic individuals, such as the A4 study.5
In the study by Jack and colleagues,4 amyloid PET imaging was performed with Pittsburgh Compound B (PiB) and tau PET with flortaucipir. Amyloid and tau PET standardized uptake value ratios (SUVRs) were formed by normalizing composite multiregion target regions of interest to the cerebellar crus gray matter. Cut points to determine normal vs abnormal studies were SUVR 1.48 (centiloid 22) for amyloid PET and SUVR 1.25 for tau PET. While the field has gained extensive experience with PiB, detailed studies on the correlation between in vivo PiB-PET uptake and quantitative measures Aβ pathology burden at postmortem, which are the most critical for the correct interpretation of PET images, continue to be scarce.6–8 Experience with flortaucipir is much more limited because tau PET imaging remains an emerging field. It has been shown that this tracer binds with strong affinity to tangles in AD and those that form as a function of age in the setting of some off-target processes as well9; however, detailed imaging-postmortem correlation studies on flortaucipir-imaged individuals are still missing, and cutoffs for flortaucipir in vivo retention have yet to be defined. These are critical aspects for considering this tau tracer as a disease- and progression-specific biomarker.
An additional challenge that needs to be addressed is that, while it is widely assumed that plaques and tangles are causally related to the cognitive symptoms in AD, observations from multiple studies suggest that associations between plaques, tangles, and cognition are not particularly strong and do not suffice to reliably predict clinical outcome at an individual level.10 Multiple other factors contribute to the wide heterogeneity of the clinical expression in the form of dementia for classic brain AD neuropathologic changes, including the high prevalence of age-related intercurrent pathologies, different burden and regional lesion distribution, inflammatory or vascular factors, heterogeneous individual profiles of risk and protective factors, overlapping clinical symptoms among syndromes, or variable interindividual rates of cognitive decline. Such heterogeneity is one of the main obstacles for the rational design of prevention clinical trials among asymptomatic individuals who harbor plaques and tangles in their brains. Moreover, although the prevalence of both plaques and tangles and dementia certainly increases with age, not everyone with brain AD pathology will experience cognitive decline during their lifetime. Even excluding individuals who die of other diseases before becoming symptomatic from the lesions in their brains, not all the older adults who are positive for amyloid and tau by in vivo imaging but are clinically unimpaired will develop dementia. There is strong evidence that some individuals may be resilient to the insult of amyloid and tau deposition in their brains.11 In this scenario, decisions regarding potential therapies might not be trivial for an asymptomatic imaging-positive individual, and it would greatly enhance the power of secondary prevention trials to be able to precisely predict which asymptomatic biomarker-positive elderly persons will develop clinical symptoms of dementia and over what time frame.
The study by Jack and colleagues4 represents a step forward in demonstrating by in vivo neuroimaging the high prevalence of age-associated amyloid and tau abnormalities among asymptomatic individuals. This is in agreement with findings from traditional large autopsy series,12 highlighting a conundrum (pathologic and biomarker findings vs clinically defined entities) that can be both confusing and troubling to patients, families, and physicians and that may require the introduction of new nomenclature for the clinical state and/or the pathologic process. The existence of a long clinically silent phase in AD poses a serious challenge for the design of outcome measures in prevention trials and drives up the cost and length of these trials, particularly in the absence of another surrogate marker that can be measured in vivo to estimate how far any given individual could be from theoretically entering a symptomatic phase. Subsequent future investigations must be directed to understanding the pathologic and biochemical brain changes, as well as individual genetic and epigenetic factors that, beyond just the presence of plaques and tangles, drive the clinical course in these imaging-positive individuals. Such work has the potential to accurately predict the future for an individual who is cognitively normal yet “imaging positive.” Understanding who and when will allow personalized assessment of the need and optimal timing for preventive intervention.